Ryan Ames r.ames@exeter.ac.uk
RNA sequencing (RNA-Seq) is a powerful method for discovering, profiling, and quantifying RNA transcripts using massively parallel sequencing technologies. These sequencing technologies are capable of generating hundreds of millions of reads, which unlike Sanger sequencing approaches; enable quantification of transcripts as well as discovery of transcripts and novel isoforms. Most sequencing technologies do not permit direct sequencing of RNA molecules. Instead RNA molecules of interest are extracted from cells of the organism of interest. The RNA is purified and converted into cDNA via reverse transcription. From the derived sequences it is possible to perform several types of analysis. RNA-seq data can be used to validate gene predictions, find novel splice variants or detect gene fusions in cancers for example; but perhaps the most common type of experiment and the one we will be looking at here, is identifying genes which are differently expressed under different conditions.
A typical sequencing run would begin with the user supplying 1-10 µg of RNA to a facility along with quality control information in the form of an Agilent Bioanalyser trace or gel image and quantification information. The following figure illustrates the basic workflow.
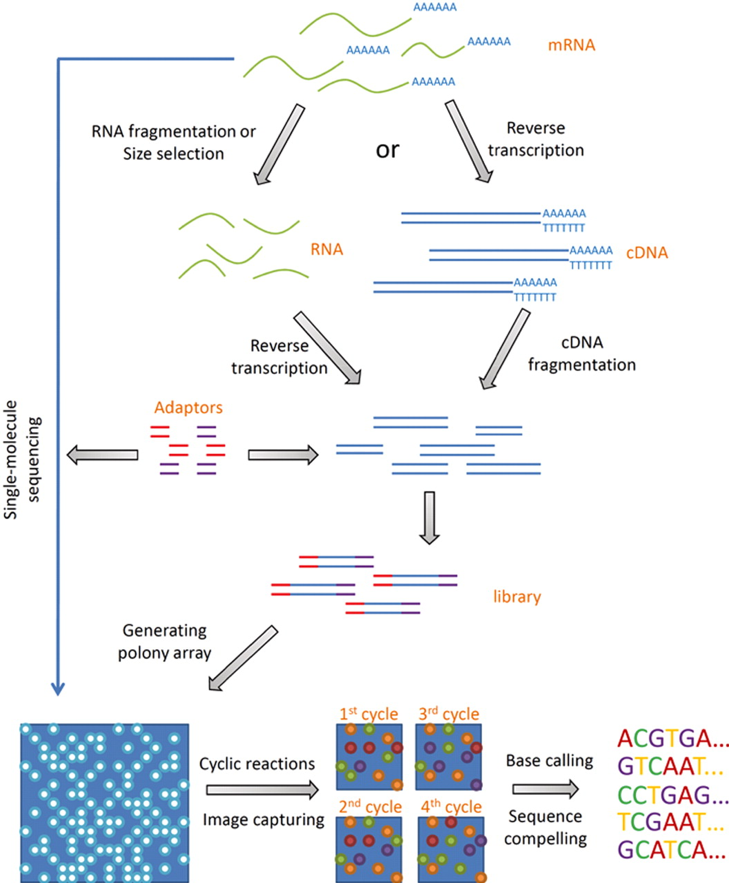
Overview of RNA-seq experimental procedures. For a typical RNA-seq experiment, mRNA is isolated and reverse-transcribed (RT) into cDNA libraries with homogeneous lengths. This is achieved by either RNA or cDNA fragmentation. Adaptors at one or both ends of the RNA are added prior to cDNA amplification and library construction. For the Illumina platform, cDNA molecules are anchored onto a flow cell surface, which are then subjected to PCR amplification. Images are taken after each cycle for base calling and sequence generation. Currently for the Illumina platform, ∼2 billion single or paired-end reads of 125nt are generated on a single flow cell (8 lanes/flow cell) which is then processed further depending on the research goals.
One important feature of RNA-seq is that the data sets are quantitative - the more highly expressed a gene is, the more frequently it will occur in the data. This can then be compared to other samples to look for significant differences in the abundance of mRNA, known as Differential Gene Expression (DGE) analysis.
For more background checkout Wang et al., 2009 and Ozsolak & Milos, 2011.
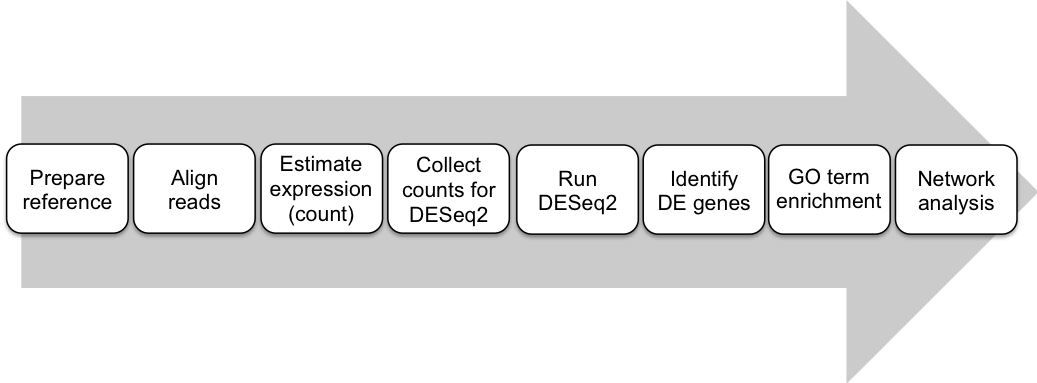
In this workshop we will look at the analysis of an RNA-seq dataset. Specifically: a differential gene expression experiment, where we attempt to find a set of genes that are expressed at different levels in two strains of Arabidopsis Thaliana, when challenged with a fungal pathogen Sclerotinia sclerotiorum. Following on from DGE analysis we will identify enriched GO terms in the set of differentially expressed genes. This will allow us to assign specific functions to these genes.
Here is the workflow that we will use in this workshop